La distrofia vitelliforme foveomaculare ad esordio tardivo, nell’età adulta, è una distrofia maculare genetica, caratterizzata da annebbiamento visivo, metamorfopsia e perdita della vista. A livello retinico si evidenzia una lesione giallognola peculiare ”a tuorlo d’uovo” leggermente rialzata, localizzata in regione foveale o parafoveale.
Epidemiologia
Questo tipo di patologia esordisce tra la quarta e sesta decade di vita. Nelle fasi iniziali i pazienti non presentano sintomi visivi o sono interessati in modo lieve, l’acuità visiva di solito rimane conservata. Però quando la malattia progredisce, la perdita della vista può aggravarsi. Possono essere presenti neovascolarizzazione coroidale (CNV) ed atrofia centrale dell’epitelio pigmentato retinico.
Diagnosi
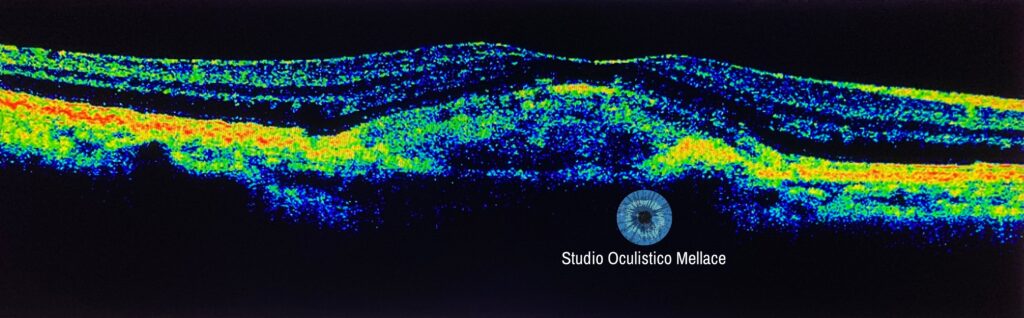
La diagnosi si basa sull’esame oftalmologico completo, sull’autofluorescenza del fondo dell’occhio, sulla tomografia a coerenza ottica e sull’angiografia con fluoresceina, nel caso si sospetti una neovascolarizzazione coroideale. L’OCT rivela una lesione vitelliforme localizzata a livello dell’EPR o tra l’EPR e i fotorecettori. Questa tecnica è estremamente utile per distinguere la distrofia vitelliforme foveomaculare dalla degenerazione maculare correlata con l’età.
Terapia
Non è disponibile una terapia efficace per la distrofia vitelliforme foveomaculare ed i pazienti dovrebbero sottoporsi ad una visita oculistica completa, una o due volte all’anno, al fine di escludere possibili complicazioni, come la CNV, i fori maculari a tutto spessore, o i distacchi della retina.
La prognosi risulta essere buona in quanto la malattia causa tipicamente una perdita della vista lenta e progressiva; la maggior parte dei pazienti conserva una vista sufficiente per molto tempo nel corso dell’evoluzione della malattia.